Systematic structure–activity relationship (SAR) studies are conducted to identify novel antiviral agents, particularly compounds active against resistant viral mutants that are insensitive to commonly used therapies. The activity of acyclic nucleoside phosphonates (ANPs), in which a nucleobase is linked to a phosphonate moiety via a suitable spacer, is based on their structural similarity to naturally occurring metabolites.
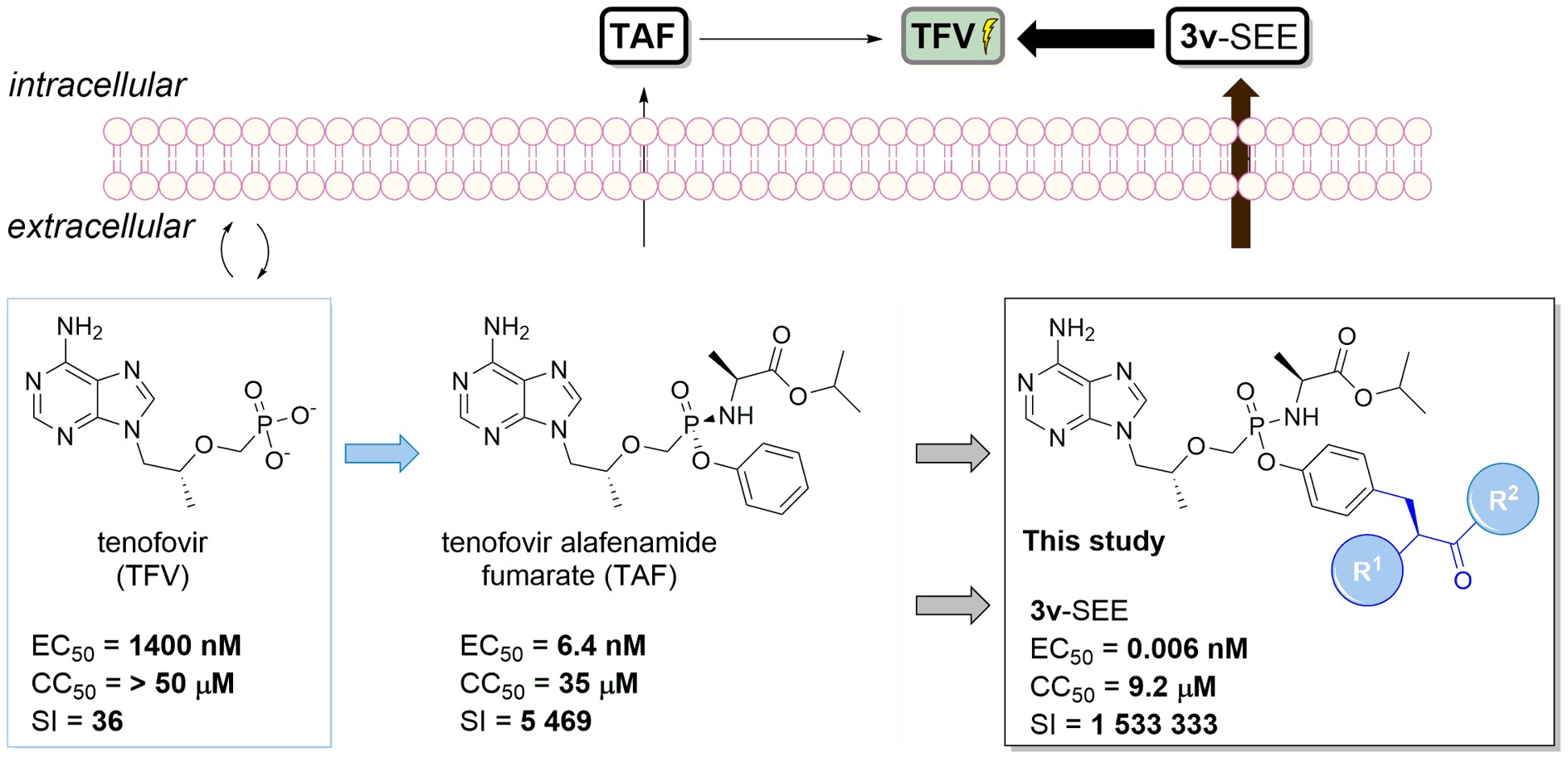
ANPs represent highly attractive lead structures for antiviral drug design due to their conformational flexibility, the absence of a labile glycosidic bond, and the enhanced enzymatic and chemical stability of the phosphonate group compared with conventional phosphate esters. A key component of ANP development is the application of prodrug strategies, which effectively overcome the intrinsic polarity of phosphonates. By masking the charged phosphonate group, prodrugs significantly improve membrane permeability, oral bioavailability, and intracellular delivery of the active compound. The success of this approach is clearly demonstrated by tenofovir and its clinically used prodrugs, tenofovir disoproxil fumarate and tenofovir alafenamide, which achieve substantially improved pharmacokinetic properties and therapeutic efficacy compared with the parent drug.
Within our research, effects of diverse structural modifications of both the nucleobase and the acyclic moiety on biological activity are systematically investigated, together with the development of novel prodrug strategies aimed at improving pharmacokinetic properties, cellular uptake, and overall therapeutic efficacy. Broad-spectrum antiviral activities are evaluated in collaboration with the Rega Institute for Medical Research at KU Leuven (Belgium) and with Gilead Sciences (USA).
Anticancer agents
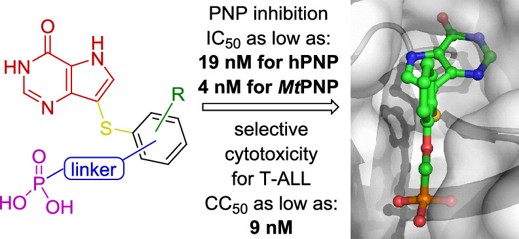
Purine nucleoside phosphorylases (PNPs) are cytosolic enzymes that play a central role in the purine salvage pathway in most organisms. They catalyze the phosphorolytic cleavage of (2′-deoxy)nucleosides derived from 6-oxopurines, producing the corresponding purine bases and (2-deoxy)ribose-1-phosphate. Despite their biological relevance, no PNP inhibitor (PNPI)-based therapy has yet been approved by the FDA or EMA (although forodesine (BCX-1777, Immucillin-H) was approved in March 2017 in Japan).
In our group, we have identified a new class of phosphonate-based PNP inhibitors. To access these compounds, we developed an efficient synthetic methodology that enables the combination of key structural motifs from several highly potent PNP inhibitors that were previously synthetically inaccessible. This approach has also significantly expanded the accessible chemical space of known PNP inhibitors. These compounds and their derivatives remain under active investigation as promising pharmacological tools and potential therapeutic leads.
In addition to this project, we are actively working on several other anticancer projects, which are at various stages of research.
Antiparasitics
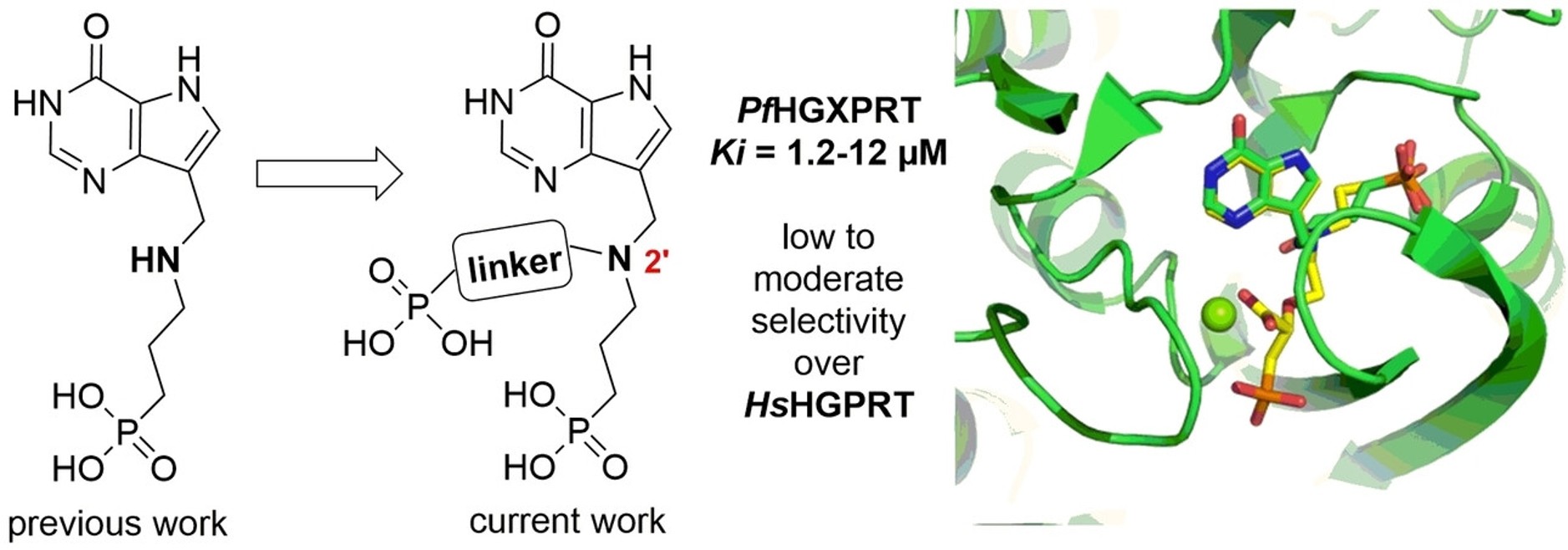
Hypoxanthine-guanine-(xanthine) phosphoribosyltransferase (HG(X)PRT) is an essential enzyme required for the survival of the malaria parasites Plasmodium falciparum and Plasmodium vivax. In contrast to plasmodia, humans possess both de novo and salvage pathways for purine biosynthesis and are therefore less dependent on the purine salvage pathway. This metabolic difference provides a strong rationale for the design of selective inhibitors of plasmodial HG(X)PRT, which could suppress parasitemia while minimizing toxicity to the human host. In advance, it has been shown that previously mentioned inhibition of PNP leads to disruption of a nucleotide pool in cells, which affects viability of various pathogenic organisms.
Recently, a novel class of aza-acyclic nucleoside phosphonates (aza-ANPs) has been designed, synthesized, and biologically evaluated. Several of these compounds exhibit low Ki values toward plasmodial PfHGXPRT and PvHGPRT enzymes together with pronounced selectivity over the human HGPRT enzyme, indicating their potential as promising lead structures for the development of new antimalarial agents. Furthermore, inhibition of human HGPRT can be exploited to fight various types of cancer. The investigation of ANP inhibitory activity is conducted in international collaborations.
Antibiotics
Anthrax toxin is a tripartite exotoxin secreted by virulent strains of Bacillus anthracis, the causative agent of anthrax. It consists of three components: protective antigen (PA), edema factor (EF), and lethal factor (LF). Edema factor is a Ca²⁺- and calmodulin-dependent adenylate cyclase that causes a marked increase in intracellular cAMP levels, thereby disrupting key host cellular processes.
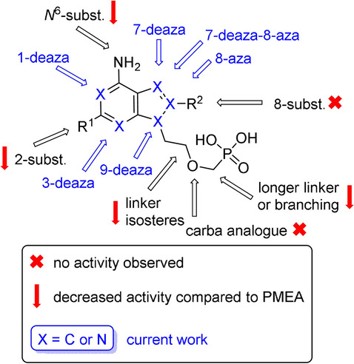
It has been demonstrated that PMEA diphosphate (PMEApp) inhibits adenylate cyclase activity both in the adenylate cyclase toxin (ACT) of Bordetella pertussis and in the edema factor of Bacillus anthracis. PMEApp, as a structural analogue of ATP, acts as a competitive inhibitor by binding to the enzyme’s active site. Within this project, we have synthesized and evaluated a range of structurally diverse PMEA analogues as potential inhibitors of adenylate cyclase. The obtained results are encouraging and support the further development of these compounds as promising leads for novel therapeutic strategies.